We Are Crinetics
Crinetics is a pharmaceutical company founded in endocrinology research. Our intense focus is on developing much-needed therapies for people with rare endocrine diseases. For us, it’s all about these underserved patients, who are eager to find therapies that provide effective disease control and bring more simplicity to their lives. To better achieve these goals, we also partner with endocrinologists and other healthcare practitioners to ensure we’re solving real problems for them.
| Discovery | Pre-Clinical | Phase 1 | Phase 2 | Phase 3 |
|---|---|---|---|---|
| Discovery | Pre-Clinical | Phase 1 | Phase 2 | Phase 3 |
|---|---|---|---|---|
Inhibition of Basal and ACTH-Stimulated Cortisol Secretion in Humans Using an Oral, Nonpeptide ACTH Antagonist (CRN04894)

CRN04777, an Oral, Nonpeptide SST5-selective Somatostatin Agonist Dose Dependently Suppresses Basal and Stimulated Insulin Secretion

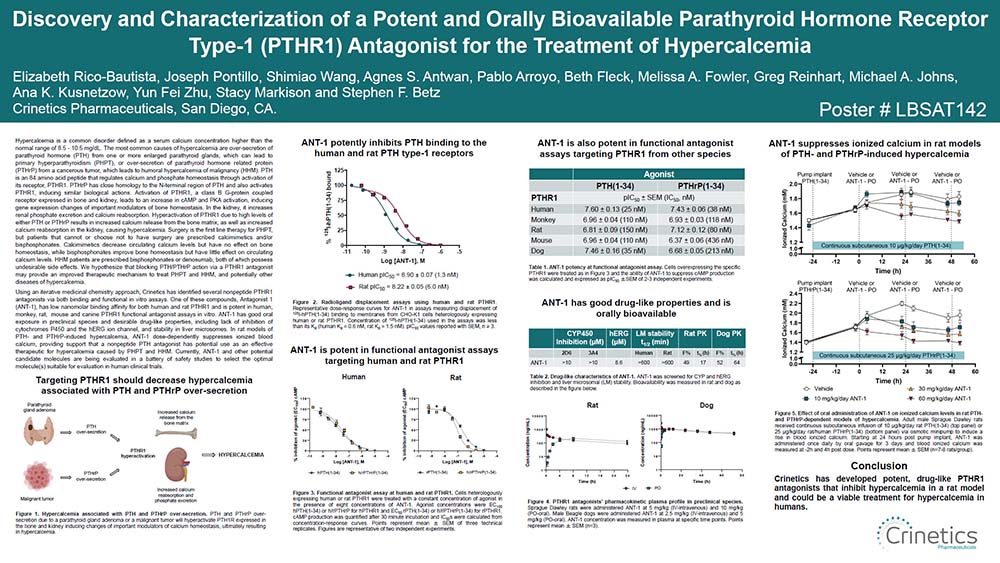
Discovery and Characterization of a Potent and Orally Bioavailable Parathyroid Hormone Receptor Type-1 (PTHR1) Antagonist for the Treatment of Hypercalcemia
Fill out this form to receive a link to download our ENDO 2022 presentations.
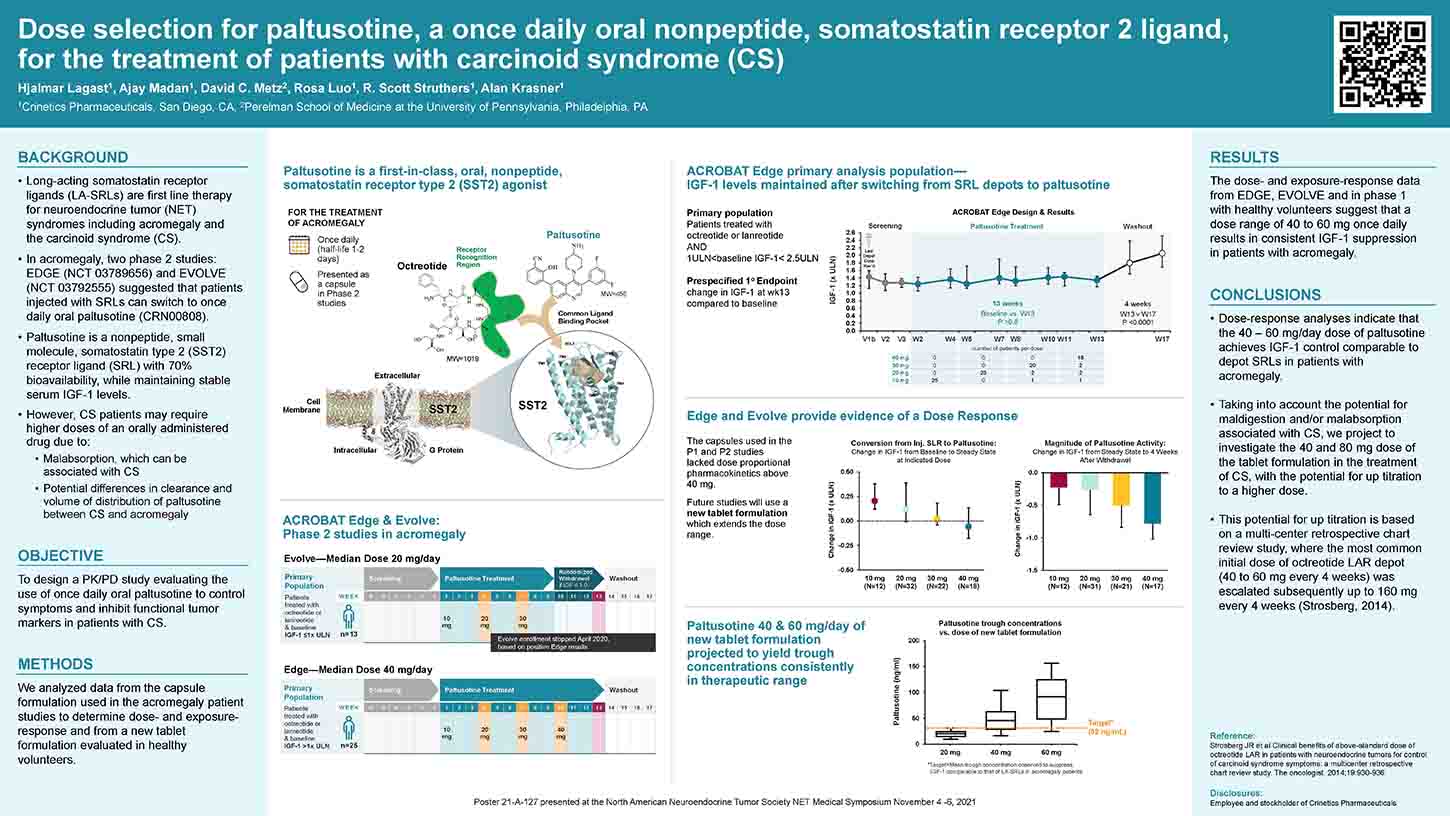
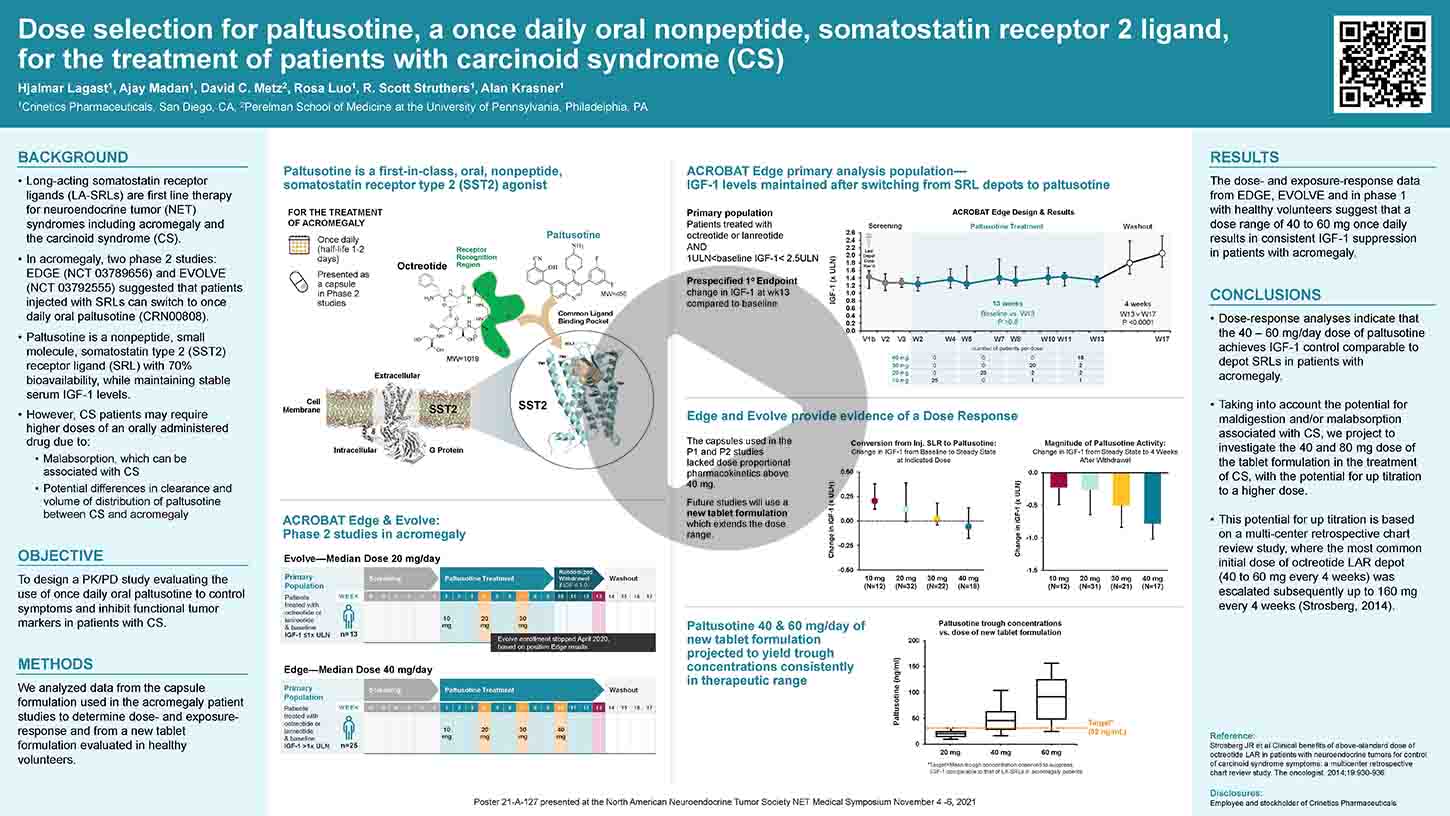
Dose selection for paltusotine, a once-daily, oral, nonpeptide, somatostatin receptor 2 ligand (SST2), for the treatment of patients with carcinoid syndrome (CS)
Pharmacokinetics and Safety of an Improved Oral Formulation of Paltusotine, a Selective, Non-Peptide Somatostatin Receptor 2 (SST2) Agonist for the Treatment of Acromegaly
Improved Oral Formulation of Paltusotine for the Treatment of Acromegaly
Authors
Rosa Luo, MS1, Gerald Burke, PhD1, Cosina Mui, BSc1, Sepehr Shakib, Prof2, Christine Ferrara-Cook, M.D., Ph.D.1, Alan Krasner, MD1, Ajay Madan, PhD1. 1Crinetics Pharmaceuticals, Inc., San Diego, CA, USA, 2CMAX Clinical Research Pty Ltd, Adelaide, Australia.
Disclosures
R. Luo: Employee; Self; Crinetics Pharmaceuticals. G. Burke: None. C. Mui: Employee; Self; Crinetics Pharmaceuticals. S. Shakib: None. C. Ferrara-Cook: Employee; Self; Crinetics Pharmaceuticals. A. Krasner: Employee; Self; Crinetics Pharmaceuticals. A. Madan: Employee; Self; Crinetics Pharmaceuticals.
Abstract
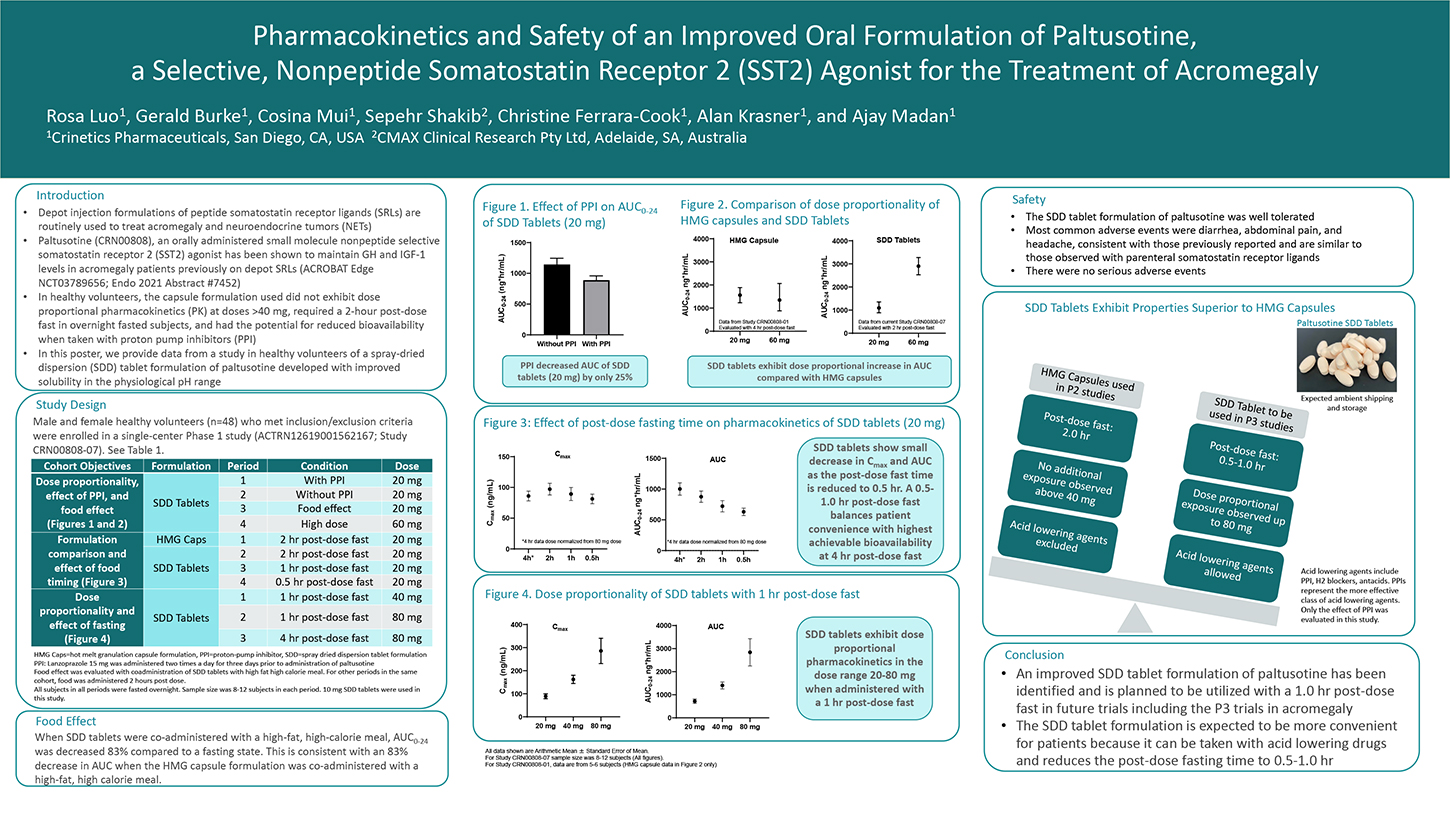
Depot injection formulations of peptide somatostatin receptor ligands (SRLs) are routinely used to treat acromegaly and neuroendocrine tumors (NETs). Paltusotine (CRN00808), an orally administered small molecule nonpeptide selective somatostatin receptor 2 (SST2) agonist has been shown to maintain GH and IGF-1 levels in acromegaly patients previously on depot SRLs (ACROBAT Edge NCT03789656). In this study, a capsule formulation was used, which did not exhibit dose proportional pharmacokinetics (PK) at doses >40 mg, required a 2-hour post-dose fast in overnight fasted patients, and had the potential for reduced bioavailability when taken with proton-pump inhibitors (PPI). A spray-dried dispersion (SDD) tablet formulation was developed with improved solubility in the physiological pH range and its performance was evaluated in healthy volunteers. Male and female healthy volunteers who met inclusion/exclusion criteria were enrolled in a single-center Phase 1 study (ANZCTR registration ACTRN12619001562167). A Cohort of 12 subjects was administered a single dose of paltusotine in a four-period cross-over design. Periods 1 and 2 assessed the effect of lansoprazole (a PPI) on PK of 20 mg dose of paltusotine SDD tablets.
In Period 3, 20 mg dose of paltusotine SDD tablets was co-administered with a high fat, high-calorie meal. In Period 4, a 60 mg dose of paltusotine SDD tablets was administered to assess dose proportionality. In a separate cohort of subjects (n=12; also, a 4-period cross- over design), the relative bioavailability of capsules and SDD tablets was assessed, and the effect of food administration 0.5, 1, and 2 hour post-dose was evaluated. Subsequently, in another cohort of 12 subjects (a 3-period cross-over design), dose proportionality of the SDD tablets was evaluated at 40 mg and 80 mg dose with a 1-hour post-dose fast. A 4 hours post-dose fast was also assessed for the 80 mg dose. Pharmacokinetics and safety of paltusotine were evaluated.
Paltusotine was generally well tolerated in this study. SDD tablets exhibited dose proportional increase in total systemic exposure (AUC) up to a dose of 80 mg. Healthy volunteers pretreated with the PPI, lansoprazole (15 mg bid for 3 days), and co-administered with paltusotine SDD tablets exhibited a small decrease (approximately 25%) in systemic exposure to paltusotine compared with the same subjects that had washed out from the PPI-pretreatment. SDD tablets exhibited significant reduction in systemic exposure when co-administered with a high-fat, high-calorie meal. However, the SDD formulation was less sensitive to timing of post-dose food administration compared to the capsule formulation. These data suggest that the SDD tablet formulation of paltusotine improves flexibility in dose administration, can be co-administered with PPIs and other agents that increase stomach pH, and reduces the post-dose fasting requirement.
1) Crinetics Pharmaceuticals, Inc., San Diego, CA, USA, and 2) CMAX Clinical Research Pty Ltd, Adelaide, Australia.
Safety and Efficacy of Switching Injected Peptide Long-Acting Somatostatin Receptor Ligands to Once Daily Oral Paltusotine: ACROBAT Edge Phase 2 Study
Results Support Switching from Injectables to Oral Paltusotine for Acromegaly


Authors
Monica R. Gadelha, MD, PhD1, Murray B. Gordon, MD2, Mirjana Doknic, MD, PhD3, Emese Mezősi, MD, PhD, DSci4, Miklós Tóth, MD, PhD, DSci5, Harpal Randeva, MBChB, FRCP, FAcadTM, PhD6, Tonya Marmon, PhD7, Rosa Luo, MS8, Michael Monahan, MBA<sup8, Ajay Madan, PhD8, Christine Ferrara-Cook, MD, PhD8, Scott Struthers, PhD8, Alan Krasner, MD8.
Disclosures
M.R. Gadelha: Advisory Board Member; Self; Ipsen, Novartis Pharmaceuticals, Crinetics. Research Investigator; Self; Crinetics, Novartis Pharmaceuticals. Speaker; Self; Novartis Pharmaceuticals, Ipsen, Pfizer,
Inc. M.B. Gordon: Research Investigator; Self; Crinetics. M. Doknic: Research Investigator; Self; Crinetics. E. Mezősi: Research Investigator; Self; Crinetics. M. Tóth: Research Investigator; Self; Crinetics. H. Randeva: Research Investigator; Self; Crinetics. T. Marmon: Consulting Fee; Self; Crinetics. R. Luo: Employee; Self; Crinetics. M. Monahan: Employee; Self; Crinetics. A. Madan: Employee; Self; Crinetics. C. Ferrara-Cook: Employee; Self; Crinetics. S. Struthers: Employee; Self; Crinetics. A. Krasner: Employee; Self; Crinetics.
Abstract
Patients with acromegaly not cured by surgery are often initially treated with injected peptide long-acting somatostatin receptor ligands (SRLs). Non-peptide small molecules can also activate the somatostatin receptor and do so with a high degree of precision for the target therapeutic receptor subtype.
Paltusotine (formerly CRN00808) is a small molecule somatostatin type 2 (SST2) receptor agonist with high oral bioavailability (70%) and pharmacokinetic profile suitable for once daily dosing. In healthy volunteers, paltusotine has been shown to lower growth hormone (GH) and insulin-like growth factor-1 (IGF-1) levels. We hypothesized that patients with acromegaly could switch from injected SRLs to once daily oral paltusotine while maintaining baseline IGF-1 levels. ACROBAT Edge (NCT03789656) was a single-arm study designed
to evaluate the safety and efficacy of switching from injected SRLs to paltusotine in patients with acromegaly. The primary analysis population consisted of those who had not achieved normal IGF-1 levels despite stable therapy with long-acting octreotide or lanreotide. Eligible patients received their last injection of SRL 4 weeks prior to switching to once daily oral paltusotine monotherapy for a 13-week treatment period. The starting dose of 10 mg per day was uptitrated in 10 mg increments at specified study visits to a maximal dose of 40 mg per day based on protocol specified study drug toleration and IGF-1 criteria.
The primary endpoint was change in IGF-1 from baseline to the completion of the 13-week treatment period. Statistical testing was based on non-parametric Wilcoxon Sign Rank test of whether the median change is different from zero. In addition, the rise in IGF-1 during a 4-week washout period was used to provide supportive evidence of efficacy. Twenty- five patients were enrolled in the primary analysis group, three patients discontinued from the study for non-study drug related reasons, two during the treatment period and one during the washout period after completing treatment. The primary endpoint was achieved as paltusotine treatment resulted in no significant change in IGF-1 levels at week 13 compared to baseline [change in IGF-1 =-0.034 (-0.107, 0.107), median (IQR), p>0.6]. Of the 23 patients who completed the dosing period, 20 (87%) achieved IGF-1 levels at the end of treatment that were within 20% of baseline or lower. Median IGF- 1 values rose significantly after paltusotine washout (p<0.0001). The most common treatment-emergent adverse events (>10%) included: headache, arthralgia, fatigue, peripheral swelling, paresthesia and hyperhidrosis. There were no discontinuations due to adverse events and no treatment related serious adverse events. These results suggest that patients with acromegaly treated with injected SRLs can switch to oral paltusotine while maintaining IGF- 1 and that paltusotine appeared to be well tolerated.
1Neuroendocrinology Research Center/Endocrinology Division – Medical School and Hospital Universitario Clementino Fraga Filho-Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil, 2Allegheny General Hospital, Pittsburgh, PA, USA, 3Clinical Center of Serbia, Belgrade, Serbia, 4University of Pécs Medical School, Pécs, Hungary, 5Semmelweis University, Budapest, Hungary, 6University Hospitals Coventry and Warwickshire NHS Trust, Coventry, United Kingdom, 7Marmon Biostatistics, Seattle, WA, USA, 8Crinetics Pharmaceuticals Inc., San Diego, CA, USA.
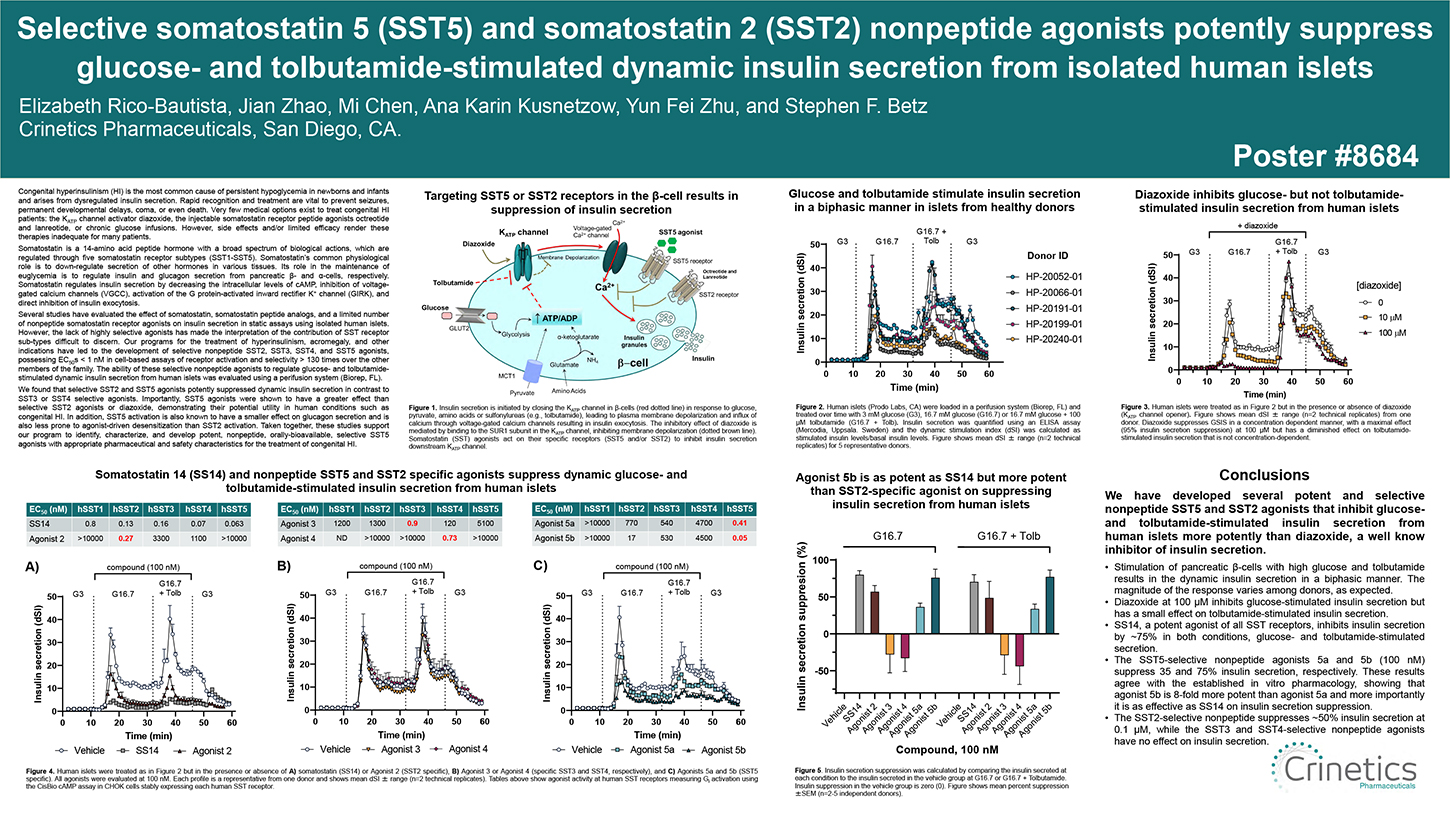
Selective Somatostatin 5 (SST5) and Somatostatin 2 (SST2) Nonpeptide Agonists Potently Suppress Glucose- and Tolbutamide-Stimulated Dynamic Insulin Secretion From Isolated Human Islets
Insulin Regulation with Selective SST5 Agonist (congenital hyperinsulinism)

Authors
Elizabeth Rico, PhD, Jian Zhao, PhD, Mi Chen, BS, Ana Karin Kusnetzow, PhD, Yun Fei Zhu, PhD, Stephen F. Betz, PhD.
Crinetics Pharmaceuticals, San Diego, CA, USA.
Disclosures
E. Rico: Employee; Self; Crinetics Pharmaceuticals. J. Zhao: Employee; Self; Crinetics Pharmaceuticals. M. Chen: Employee; Self; Crinetics Pharmaceuticals. A.K. Kusnetzow: Employee; Self; Crinetics Pharmaceuticals. Y. Zhu: Employee; Self; Crinetics Pharmaceuticals. S.F. Betz: Employee; Self; Crinetics Pharmaceuticals.
Abstract
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in newborns and infants and arises from dysregulated insulin secretion. Rapid recognition and treatment are vital to prevent seizures, permanent developmental delays, coma, or even death. Very few medical options exist to treat congenital HI patients: the KATP channel activator diazoxide, the injectable somatostatin receptor peptide agonists octreotide and lanreotide, or chronic glucose infusions. However, side effects and/or limited efficacy render these therapies inadequate for many patients.
Somatostatin is a 14-amino acid peptide hormone with a broad spectrum of biological actions, which are regulated through five somatostatin receptor subtypes (SST1-SST5). Somatostatin’s common physiological role is to down- regulate secretion of other hormones in various tissues. Its role in the maintenance of euglycemia is to regulate insulin and glucagon secretion from pancreatic β- and α-cells, respectively. Somatostatin regulates insulin secretion by decreasing the intracellular levels of cAMP, inhibition of voltage-gated calcium channels (VGCC), activation of the G protein-activated inward rectifier K+ channel (GIRK), and direct inhibition of insulin exocytosis.
Several studies have evaluated the effect of somatostatin, somatostatin peptide analogs, and a limited number of nonpeptide somatostatin receptor agonists on insulin secretion in static assays using isolated human islets.
However, the lack of highly selective agonists has made the interpretation of the contribution of SST receptor sub-types difficult to discern. Our programs for the treatment of hyperinsulinism, acromegaly, and other indications have led to the development of selective nonpeptide SST2, SST3, SST4, and SST5 agonists, possessing EC50s < 1 nM in cell-based assays of receptor activation and selectivity > 130 times over the other members of the family. The ability of these selective nonpeptide agonists to regulate glucose- and tolbutamide- stimulated dynamic insulin secretion from human islets was evaluated using a perifusion system (Biorep, FL).
We found that selective SST2 and SST5 agonists potently suppressed dynamic insulin secretion in contrast to SST3 or SST4 selective agonists. Importantly, SST5 agonists were shown to have a greater effect than selective SST2 agonists or diazoxide, demonstrating their potential utility in human conditions such as congenital HI. In addition, SST5 activation is also known to have a smaller effect on glucagon secretion and is also less prone to agonist-driven desensitization than SST2 activation. Taken together, these studies support our program to identify, characterize, and develop potent, nonpeptide, orally-bioavailable, selective SST5 agonists with appropriate pharmaceutical and safety characteristics for the treatment of congenital HI.
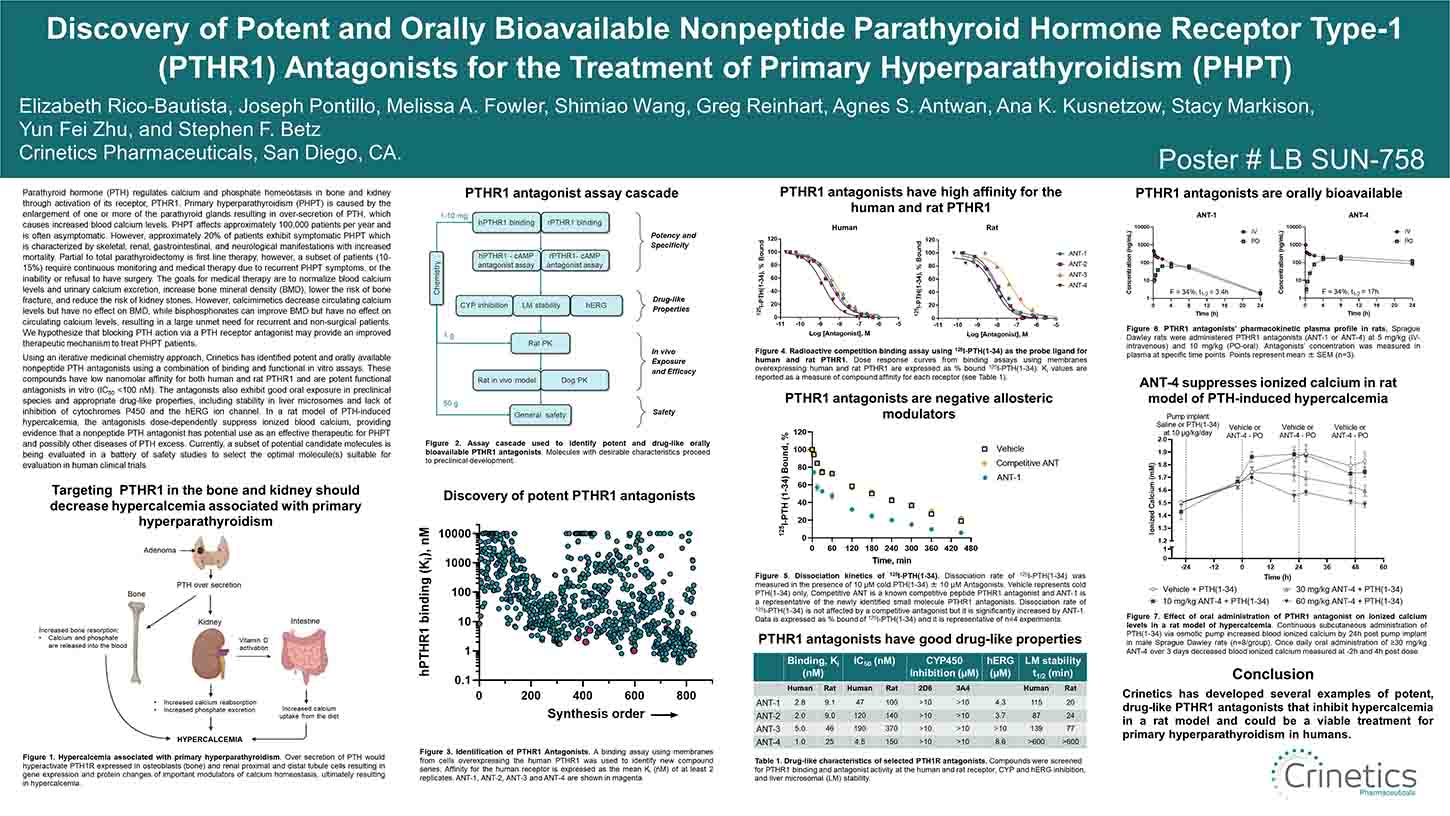
Discovery of Potent and Orally Bioavailable Nonpeptide Parathyroid Hormone Receptor Type-1 (PTHR1) Antagonists for the Treatment of Primary Hyperparathyroidism (PHPT)
Authors
Elizabeth Rico-Bautista, Joseph Pontillo, Melissa A. Fowler, Shimiao Wang, Greg Reinhart, Agnes S. Antwan, Ana K. Kusnetzow, Stacy Markison, Yun Fei Zhu, and Stephen F. Betz
Crinetics Pharmaceuticals, San Diego, CA.
Disclosures
E. Rico-Bautista: Employee; Self; Crinetics Pharmaceuticals. J. Pontillo: Employee; Self; Crinetics Pharmaceuticals. M.A. Fowler: Employee; Self; Crinetics Pharmaceuticals. S. Wang: Employee; Self; Crinetics Pharmaceuticals. G. Reinhart: Employee; Self; Crinetics Pharmaceuticals. A.S. Antwan: Employee; Self; Crinetics Pharmaceuticals. A.K. Kusnetzow: Employee; Self; Crinetics Pharmaceuticals. S. Markison: Employee; Self; Crinetics Pharmaceuticals. Y.F. Zhu: Employee; Self; Crinetics Pharmaceuticals. S.F. Betz: Employee; Self; Crinetics Pharmaceuticals.
Abstract
Parathyroid hormone (PTH) regulates calcium and phosphate homeostasis in bone and kidney through activation of its receptor, PTHR1. Primary hyperparathyroidism (PHPT) is caused by the enlargement of one or more of the parathyroid glands resulting in over-secretion of PTH, which causes increased blood calcium levels. PHPT affects approximately 100,000 patients per year and is often asymptomatic. However, approximately 20% of patients exhibit symptomatic PHPT which is characterized by skeletal, renal, gastrointestinal, and neurological manifestations with increased mortality. Partial to total parathyroidectomy is first line therapy, however, a subset of patients (10-15%) require continuous monitoring and medical therapy due to recurrent PHPT symptoms, or the inability or refusal to have surgery. The goals for medical therapy are to normalize blood calcium levels and urinary calcium excretion, increase bone mineral density (BMD), lower the risk of bone fracture, and reduce the risk of kidney stones. However, calcimimetics decrease circulating calcium levels but have no effect on BMD, while bisphosphonates can improve BMD but have no effect on circulating calcium levels, resulting in a large unmet need for recurrent and non-surgical patients. We hypothesize that blocking PTH action via a PTH receptor antagonist may provide an improved therapeutic mechanism to treat PHPT patients.
Using an iterative medicinal chemistry approach, Crinetics has identified potent and orally available nonpeptide PTH antagonists using a combination of binding and functional in vitro assays. These compounds have low nanomolar affinity for both human and rat PTHR1 and are potent functional antagonists in vitro (IC50 <100 nM). The antagonists also exhibit good oral exposure in preclinical species and appropriate drug-like properties, including stability in liver microsomes and lack of inhibition of cytochromes P450 and the hERG ion channel. In a rat model of PTH-induced hypercalcemia, the antagonists dose-dependently suppress ionized blood calcium, providing evidence that a nonpeptide PTH antagonist has potential use as an effective therapeutic for PHPT and possibly other diseases of PTH excess. Currently, a subset of potential candidate molecules is being evaluated in a battery of safety studies to select the optimal molecule(s) suitable for evaluation in human clinical trials.
Effects of CRN04894, a Nonpeptide Orally Bioavailable ACTH Antagonist, on Corticosterone in Rodent Models of ACTH Excess
CRN04894 Reduced Effects of Excess ACTH in Animal Model of Cushing’s Disease

Authors
Melissa A. Fowler, PhD, Ana Karin Kusnetzow, PhD, Sangdon Han, PhD, Greg Reinhart, BS, Sun Hee Kim, PhD, Michael Johns, BS, Taylor A. Kredel, BS, Agnes Antwan, BS, Jon Athanacio, BS, Oleg Tsivkovski, BS, Rosa Luo, MS, Ajay Madan, PhD, Yun Fei Zhu, PhD, Stephen F. Betz, PhD, Scott Struthers, PhD, Stacy Markison, PhD. Crinetics Pharmaceuticals, San Diego, CA, USA.
Disclosures
M.A. Fowler: Employee; Self; Crinetics Pharmaceuticals. A.K. Kusnetzow: Employee; Self; Crinetics Pharmaceuticals. S. Han: None. G. Reinhart: Employee; Self; Crinetics Pharmaceuticals. S. Kim: Employee; Self; Crinetics Pharmaceuticals. M. Johns: Employee; Self; Crinetics Pharmaceuticals. T.A. Kredel: Employee; Self; Crinetics Pharmaceuticals. A. Antwan: Employee; Self; Crinetics Pharmaceuticals. J. Athanacio: Employee; Self; Crinetics Pharmaceuticals. O. Tsivkovski: Employee; Self; Crinetics Pharmaceuticals. R. Luo: Employee; Self; Crinetics Pharmaceuticals. A. Madan: Employee; Self; Crinetics Pharmaceuticals. Y. Zhu: Employee; Self; Crinetics Pharmaceuticals. S.F. Betz: Employee; Self; Crinetics Pharmaceuticals. S. Struthers: Employee; Self; Crinetics Pharmaceuticals. S. Markison: Employee; Self; Crinetics Pharmaceuticals.
Abstract
CRN04894 is an orally administered nonpeptide that is a potent and selective antagonist for adrenocorticotropic hormone (ACTH) acting at the melanocortin 2 receptor (MC2R) and is currently under development for the treatment of diseases of ACTH excess such as Cushing’s disease, congenital adrenal hyperplasia, and ectopic ACTH-secreting tumors. Cushing’s disease results from an adenoma derived from pituitary corticotropic cells that secrete excess ACTH, whereas ectopic ACTH syndrome arises from nonpituitary ACTH secreting tumors. Congenital adrenal hyperplasia is a genetic disease that results in cortisol deficiency leading to high levels of ACTH and adrenal androgens. Each of these indications is characterized by high ACTH levels that act on MC2R expressed in the adrenal cortex to drive pathological elevations of adrenally derived steroid hormones. CRN04894 blocks the action of ACTH at MC2R, providing a potential novel treatment for these diseases. Preclinical models of chronic hypercortisolemia include implantation of ACTH-secreting pituitary tumor cells in mice and continuous administration of ACTH via subcutaneously implanted osmotic pumps in rats. These models induce
features consistent with human diseases of ACTH excess including hypercortisolemia and hypertrophy of the adrenal glands. We employed both rodent models to examine the pharmacodynamic effects of CRN04894 on corticosterone levels and adrenal gland morphology. In the mouse pituitary tumor model, subcutaneous inoculation of the ACTH-secreting mouse pituitary tumor cell line, AtT-20, into immunodeficient mice resulted in formation of tumors and increased plasma ACTH and corticosterone levels. Repeated daily oral administration of CRN04894 for 14 days dose-dependently and robustly suppressed plasma corticosterone levels in mice with AtT-20 tumors. In the rat model, subcutaneous implantation of osmotic pumps delivering ACTH resulted in increased corticosterone levels, reduction in body weight, and hypertrophy of the adrenal glands after 7 days. Daily oral administration of CRN04894 over 7 days dose-dependently suppressed corticosterone levels, mitigated the effect of ACTH excess on body weight, and rescued the adrenal gland hypertrophy. These findings provide evidence that CRN04894 functions as an effective ACTH antagonist at MC2R to suppress adrenal corticosterone secretion in both mouse and rat models of ACTH excess and hypercortisolemia, thus providing a strong rationale for its potential therapeutic utility in diseases of ACTH excess. This work was supported in part by an SBIR grant from the NIH awarded to Dr. Struthers (R43- DK115245)
Connect With Us
If you’d like to connect with someone at Crinetics, or if you’d like to join our email list to receive periodic news and updates, please fill out and submit this form. We do not sell or share your information with third parties.